Analytics: Mass Spectrometry
The Challenges and Technological Opportunities in Oligonucleotide Therapeutics
Recent developments in gene therapy and the approval of mRNA vaccines for treating diseases has seen the biopharmaceutical industry’s need for more well-characterised RNA and oligonucleotides than ever before. How are more nucleic acid sequences being analysed by mass spectrometry (MS) through the development of new technologies, and how can innovations in software process data fully automate the workflow of the confirmation of oligonucleotide sequences based on intact MS/MS data, as well as identify the synthesis of the byproducts or impurities as an additional output?
Detlev Suckau and Stuart Pengelley at Bruker Daltonics
The biopharmaceutical industry’s demand for well-characterised ribonucleic acid (RNA) and oligonucleotides has soared as a result of the recent developments in gene therapy and the licensure of messenger RNA (mRNA) vaccines in treating the diseases of today. To meet this increased demand, scientific researchers have created new mass spectrometry (MS) based tools and technologies for the analysis of nucleic acid sequences. Software developments now enable the entirely automatic validation of sequences based on intact mass and tandem MS (MS/MS) data, as well as the quantification and identification of synthesis by-products. This is done by using data from high-isotopic fidelity quadrupole time-of-flight (QTOF) and trapped ion mobility mass spectrometry time-of-flight (tims-TOF) instruments. RNA and oligonucleotide sequence confirmation requires robustness, sensitivity and a broad dynamic range, all of which have been made possible by advances and innovation in analytical instrumentation.
Through this innovation, these high-performance scientific instruments can automate the validation of oligonucleotide sequences, utilising MS/MS data sets and the quantification of by-products with liquid chromatography with ultraviolet detection (LC-UV) or liquid chromatography mass spectrometry (LC-MS) from the same data set. It has been demonstrated that they permit the study of complex oligonucleotide sequences, a process that was previously time-consuming and restricted to specialists with prior knowledge. In addition, any lab-specific sequence syntaxes are supported, which facilitates the incorporation of the software into existing laboratory workflows. For the analysis of highly modified RNA, which is frequently utilised because of its increased stability over canonical RNA, the combination of isotopic fidelity and ultra-high-resolution MS has shown great potential.
The increasing significance of oligonucleotides in research, diagnostics and gene therapy, including transfer RNA (tRNA), small interfering RNA (siRNA) and other modified oligos up to 120 bases, has spurred interest in creating new methods to verify sequences and discover and quantify related contaminants. LC-UV detection and liquid chromatography tandem mass spectrometry (LC-MS/MS) are the favoured techniques for characterising highly modified oligonucleotide sequences. QTOF mass spectrometers are commonly employed for this purpose due to their ability to precisely determine the monoisotopic masses of intact oligonucleotides and the high masses of their fragment ions.
“The straightforward VIP-HESI’s operating settings and efficient active exhaust make it the easiest system to upgrade from conventional sources”

Figure 1. Extracted ion mobilogram traces of charge state 6- for original 10mer oligonucleotide and isobaric clipping variants. The measured collisional cross section of clipping variants allows clear differentiation of the two molecules
Improved data quality from the high-isotopic fidelity of QTOF and tims-TOF instrumentation has increased the demand for analytical software with better capabilities for automating sequence confirmation based on intact mass and MS/MS data, as well as the quantification and identification of synthesis by products. This approach has the potential to simplify the interpretation of data, by allowing user-defined sequence definitions to easily include the vast array of modified nucleotides often employed by the biopharmaceutical industry.
Biopharmaceutical Challenges
The sequence fidelity of RNA and oligonucleotides is a crucial factor in influencing molecular safety profiles to minimise off-target effects or toxicity risks, as well as compound efficacy to ensure correct activity and expression. Next-generation sequencing technologies are useful for oligonucleotides and RNA molecules containing canonical nucleotides, but they do not provide a full evaluation of highly modified RNA-based therapeutics. Furthermore, oligonucleotide by products may include failed nucleotide additions, nucleotide variants and cytidine-to-uridine conversions with no or +1 Da variances in molecular weight from the target product sequence, respectively. Information at the nucleotide level is required to improve oligonucleotide sequence confirmation, as it might be difficult to determine what these are using their intact mass alone.
Methods based on LC-MS/MS are well-established techniques for the oligonucleotide characterisation process. Still, the complexity of oligonucleotide MS/MS spectra sometimes poses a difficulty for the implementation of the LC-MS/MS approach. Manual interpretation of spectra is time-consuming and requires specialised analytical skills, making the complete analysis of multiple samples per day in a regular laboratory challenging and costly.
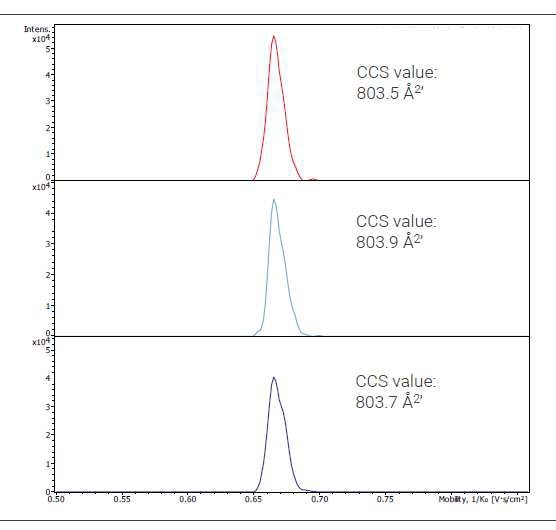
Figure 2. Extracted ion mobilogram traces of three different injections of the 3' clipped molecule. Shown is charge state 6-. Highly consistent CCS values without need of data file recalibration

Figure 3. Improved signal intensities with the use of VIP-HESI technology
Technological Opportunities
Several advances in analytical software and instrumentation hold out hope for fully automated confirmation of oligonucleotide sequences based on intact mass and MS/MS data, as well as detection and quantification of synthesised by products. With these developments in commercial software and scientific analytical instruments, RNA research and development can be expedited and accomplished with greater ease.
Trapped ion mobility spectrometry (TIMS) is primarily a gas phase separation technique that resolves sample complexity by adding a dimension of separation to high-performance liquid chromatography (HPLC) and MS, which, as a result, enhances peak capacity and confidence in compound characterisation. Importantly, the TIMS device also serves to gather and concentrate ions of a particular mass and mobility, allowing for a unique increase in sensitivity and speed as well as an extra dimension of separation. By analysing the collisional cross sections (CCS) of the two isobaric oligonucleotide clipping products with LC-TIMS-MS, it is capable of distinguishing the molecules based on an intrinsic biophysical feature. As these characteristics are independent of the analytical setup, this simplifies the method transfer.
Two isobaric 9mer oligonucleotides were investigated as an example. These two forms result from the clipping of a 10mer molecule with guanine as the base at both ends. Two isobaric 9mer oligonucleotides are produced by deleting a guanosine nucleotide from either the 5’- or 3’-terminal position. The samples were measured by HPLC using a short gradient for desalting and a mass spectrometer with TIMS functionality for differentiation based on the gas-phase behaviour features of the compounds. Extracted Ion Mobilogram (EIM) traces were created for each oligonucleotide’s measured charge states. The CCS values of the identified charge states were then computed to validate the possibility of differentiating these isobaric molecules (Figures 1 and 2, page 11).
“Manual interpretation of spectra is time-consuming and requires specialised analytical skills, making the complete analysis of multiple samples per day in a regular laboratory challenging and costly”

Figure 4. Deconvoluted spectrum for a sgRNA and associated low level impurities
“ULC-UV detection and liquid chromatography tandem mass spectroscopy (LC–MS/MS) are the favoured techniques for characterising highly modified oligonucleotide sequences”
Compared to standard electrospray, the introduction of the vacuum insulated probe (VIP) heated electrospray ionisation (HESI) ion source provides a substantial increase in sensitivity for a number of compounds of interest. The VIP-HESI provides sensitivities comparable to those of industry-leading triple-quad technology for a wide range of compounds while maintaining the well-established advantages of accurate mass values with precise isotopic patterns (TIP). The straightforward VIP-HESI’s operating settings and efficient active exhaust make it the easiest system to upgrade from conventional sources.
The VIP-HESI ion source increases sensitivity and improves detection limits across a broad range of applications by delivering more efficient ionisation. Inside the VIP-HESI, a controlled vaporising temperature is maintained for the electrospray in order to provide increased desolvation of analyte ions, even at high eluent flow rates in ultra-HPLC separations with sharp chromatographic peaks (Figure 3). Within the VIP, the eluent stream containing the target compounds is protected from heat by a vacuum barrier. The combination of heating and an eluent stream into the probe makes the design simple and easy to maintain. Using the Venturi effect, the active exhaust prevents recirculation, demonstrating a clever application of long-known physical concepts in modern analytical instrumentation.

Figure 5. Example of RNA 24mer analysis
Adding powerful tools for interpreting RNA and oligonucleotide data, the MS/MS data gap is actively being closed, yielding better outcomes and results.1The development of a more adaptable sequence editor offers a broad range of natural and synthetic nucleic acid building blocks. User-defined sequence nomenclature eliminates the need to convert laboratory-specific sequence definitions to a proprietary software format. Proven algorithms for peak detection and deconvolution of MS and MS/MS data obtained with high-isotopic fidelity ensure correct mass assignments even for samples with a high dynamic range (Figure 4).
Specifically, one proprietary algorithm has been used successfully with top-down MS to interpret spectra, including a large number of overlapping ions with multiple charges. The MS/MS annotation engine can automatically compute and identify terminal and internal fragments, enabling the speedy and intuitive interpretation of complex MS/MS data. The software uses a proprietary algorithm technique to deisotope and annotate the MS/MS data as an output. Advanced peak selection assists in the interpretation of MS/MS data containing complicated and overlapping peaks. Matching peaks are annotated directly on the spectrum (Figure 5). Fragments with multiple explanations are labeled on the spectrum with the number of matches and do not contribute to sequence coverage calculations.
The increased presence of internal fragments in MS/MS data is a common difficulty with longer molecules. Due to the limited number of possible permutations between the basic building blocks, the resulting mass degeneracy makes interpretation challenging. Internal fragments that can be annotated with a unique explanation are presented in a graphical map view to supplement the information gained from terminal fragments (Figure 6). The delta mass between internal fragments enables the confirmation of nucleotides and sequences that could not be confirmed by terminal fragments alone.
Combining all three technological innovations has created a robust and reliable analytical platform compatible with proven HPLC methods and boosted sensitivity due to optimal ionisation in the VIP-HESI ion source (Figure 7). The combination of isotopic fidelity and high resolution enables the sub-ppm mass accuracy determination of biopolymers in the 5-50 kDa range. Robustness, sensitivity and, most significantly, a high dynamic range in negative mode permit the detection and identification of contaminants based on intact mass. These properties have been demonstrated to be advantageous for the characterisation of analytes including siRNA APIs, single guide RNA (sgRNA) for gene editing and mRNA-specific tests (5’ capping, poly(A) tail).

Figure 6. Example output of unique internal fragments plus verification of the selected fragment in the profile MS/MS spectrum

HPLC separation
- Reverse phase ion pairing
MS analysis with VIP-HESI
- Ultrahigh resolution
- Targeted MS/MS or parallel accumulationserial fragmentation (PASEF)
Peak picking and deconvolution
- SNAP deisotoping for sub-ppm intact mass determination
- SNAP analysis of MS/ MS data
Data annotation with OglioQuest
- Relative quantitation based on MS1
- Annotation of fragments in MS/MS spectra
“Proven algorithms for peak detection and deconvolution of MS and MS/MS data obtained with high-isotopic fidelity ensure correct mass assignments even for samples with a high dynamic range”
Conclusion
In just a few years, oligonucleotide therapeutics have emerged from the periphery as a therapeutically relevant mechanism, due to clinical successes and the establishment of huge therapeutic pipelines benefiting from their ability to directly influence protein expression. Because high-fidelity analysis of macromolecular oligonucleotides is a fundamental building block of gene therapy, vaccines and diagnostics, it has emerged as a promising tool against the diseases of today. Improving workflows through the application of new technologies to address bottlenecks in synthesis and data interpretation will only continue to rise to meet the stringent demands and momentum of the research field and manufacturing.
References:

Detlev Suckau is Global Biopharmaceutical Solutions R&D Manager at Bruker Daltonics. He is responsible for the development of new analytical workflows and software utilising Bruker TOF and timsTOF mass spectrometers. Current development focus comprises methods for synthetic oligonucleotides characterisation and design of the software support in BioPharma Compass. He graduated in Chemistry from the University of Constance, Germany, in 1991, developing new methods to characterise the surface topology of proteins by MS. He spent a Post Doctorate year at Cornell University, US, with Fred McLafferty, analysing protein conformations in the gas phase of an FTMS, and a year in the Boehringer Mannheim Analytics Department for pharmaceutical drug development. He joined Bruker Daltonic in 1994 and has led the analytical methods developments group for more than 20 years. He has published or co-authored more than 60 peer-reviewed papers and is co-inventor of 17 patents.

Stuart Pengelley completed his Bachelor of Science in Infectious Diseases at University of East London, UK, going on to do a PhD in Virology, Reading University, UK. His Postdoc was in Mass Spectrometry at the Proteome Center Tübingen, Germany. Stuart has been at Bruker Daltonics since 2009, working in Biopharma Application Development.
Innovations in Pharmaceutical Technology (IPT)
IPT provides a platform for cutting-edge ideas, concepts, and developments shaping the future of pharmaceutical R&D.