Manufacturing: Biomanufacturing Operations
Error Mitigation in Pharmaceutical Quality Assurance and Control
Ensuring pharma products are manufactured in a safe and controlled way is a priority. How can the industry ensure that this happens?
Stephanie Harden at Waters, and Phil Nethercote (Independent Industry Consultant)
Pharmaceutical companies must continuously work towards maintaining standards for effective analytical methods and data quality to support product quality assurance, quality control, and ultimately, patient safety.
The complexity and number of variables in modern analytical test procedures increases the probability of errors; managing the response to these errors can make a large difference in the long-term impact and costs to an organisation.
One of the most significant consequences of managing such errors incorrectly are FDA warning letters/483 observations. These 483 observations are used by the FDA to communicate concerns and document observations made by FDA representatives during the inspection of a pharmaceutical or life sciences facility. Receiving a warning letter/483 observation may significantly impact a company’s reputation and public image.
However, there are proactive actions that can be taken to mitigate the risk of errors occurring and help avoid regulatory sanctions, including improved planning, management, training, workflow-building, and investment in support services. Here we outline some mitigation strategies used by pharma companies to minimise the risk of such errors.
Categorising Sources of Analytical Errors
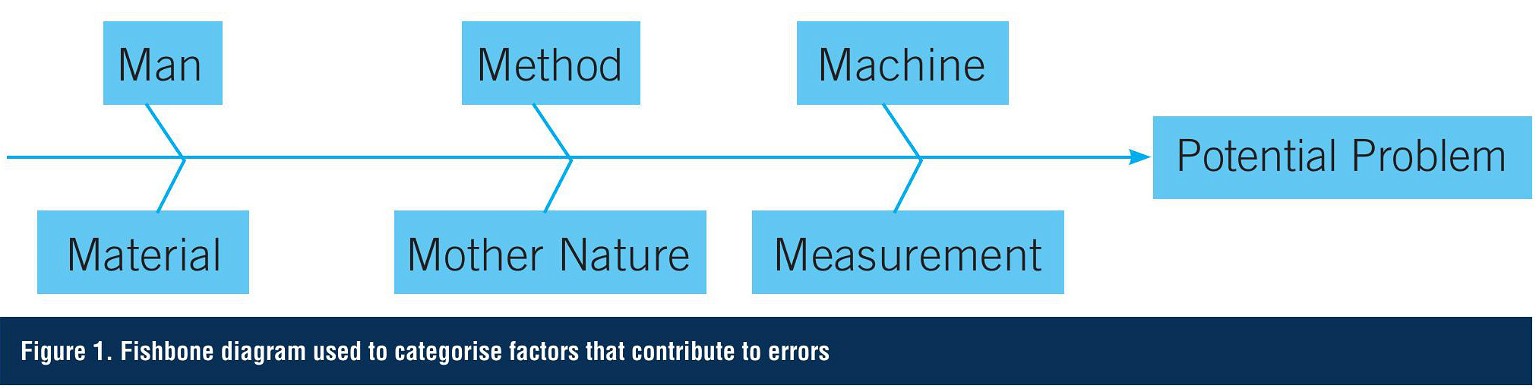
One approach used in both manufacturing and analytical testing to classify potential errors is to create a fishbone or Ishikawa diagram (after Professor Kaoru Ishikawa of Tokyo University who invented it in 1968). This methodology helps to stimulate thinking when developing the list of causes or errors. The categories often used to help teams identify factors that contribute to variations include man, method, machine, material, measurement, and Mother Nature (Figure 1).
Errors Due to Methodology
The validity of analytical test methods has traditionally been proven via well-controlled validation exercises at the end of the method development process. However, traditional approaches typically don’t include risk assessments of the procedure, and knowledge obtained under actual conditions of use can be lost. By contrast, analytical quality by design (AQbD) is an approach to method development that provides a systematic framework for continuous, real-time, and robust validation and verification. AQbD starts with predefined objectives and provides rational understanding of the effects of multiple variables on the method performance. This knowledge is used to establish the method control strategy. The outcome of this approach is a fit-for-purpose, well-designed, well-understood, and robust method that delivers the expected performance throughout its lifecycle, and significantly reduces errors due to ‘method’.
Regardless of the quality of the method development process, errors can occur if the analytical method is not executed correctly. While errors in execution may arise from any of the other areas in Figure 1, in most cases they occur due to either human error (man), instrumentation (machine), or materials. Speculations vary that anywhere from 30-80% of errors in manufacturing involve some form of human error, and similar percentages can be expected to be found within laboratories. By focusing on identifying and addressing errors due to human factors, some pharma companies have seen a 30-40% reduction in error rates.
Errors Due to Man
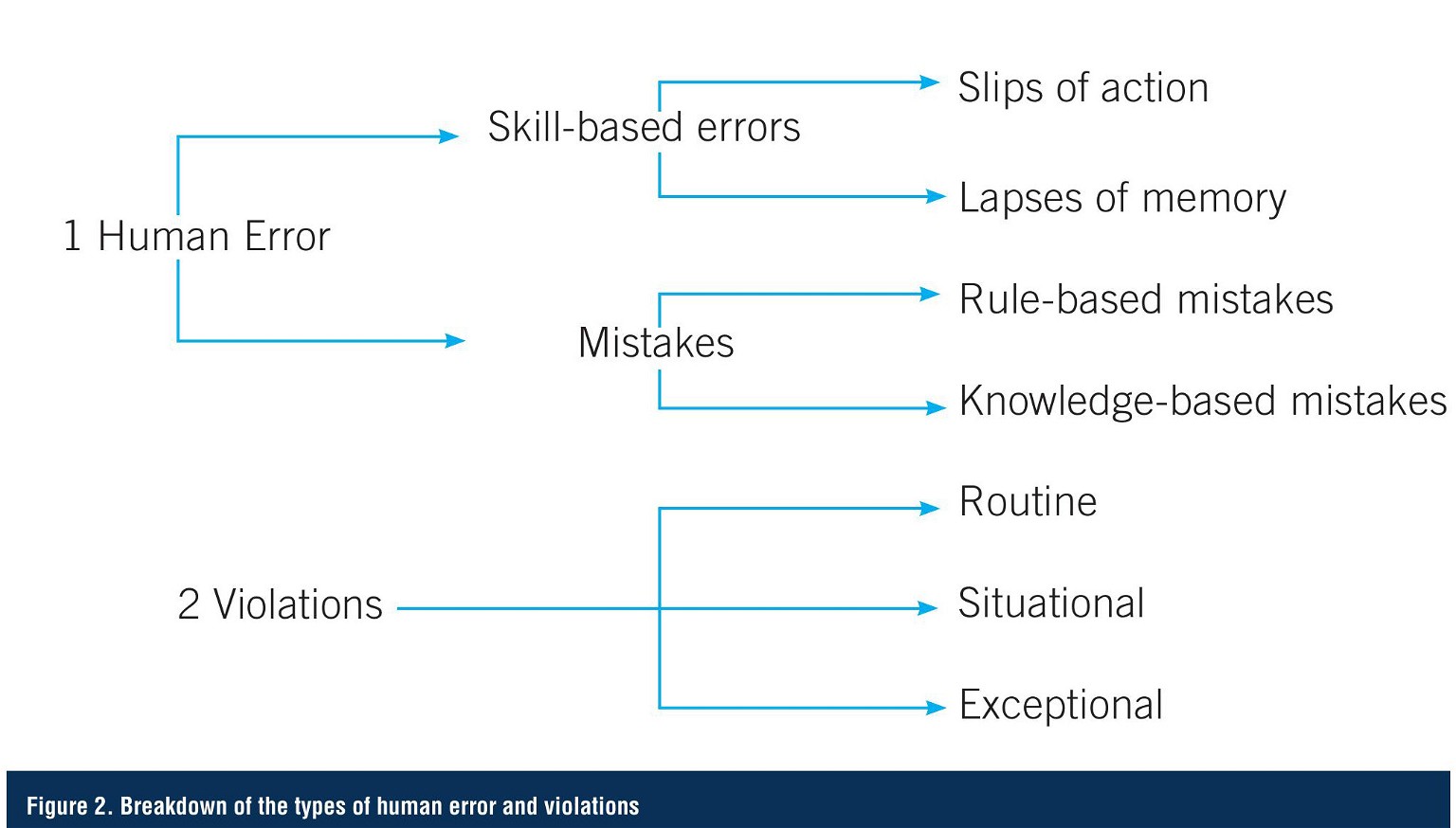
Human failure can be due to either human error or violations. Human error is an unintentional action or decision, which is caused by either a mistake, or a slip or lapse, due to a lack of required skills. Mistakes are decision-making failures – either rule-based or knowledge-based – and arise when someone does the wrong thing, believing it to be right. Common human errors in a pharma lab setting could include spills, memory lapse, lack of skill or ability, or lack of knowledge to make appropriate decisions in a non-routine situation.
In contrast, a violation is an intentional failure that was done deliberately (or with prior knowledge). Intentional actions fall into three categories: routine, situational, and exceptional. Respectively, these refer to: situations where non-compliance becomes the norm; situation-specific non-compliance; and a one-off situation where the non-compliance occurs under highly unusual circumstances. These errors might include purposely not completing a task as instructed, failing to do the task in a timely manner, or attempting to correct a mistake (Figure 2).
Mitigation Strategies
Given the frequency of errors due to ‘man’, effective strategies to prevent such issues offer significant benefits in terms of costs, time savings, and public image. Simple preventive measures based on a company’s investigation of predictable human errors can significantly reduce their occurrence. For example, if an error was due to slips or lapses in memory, ways to improve reduction in the percentage include:
- Removing distractions and interruptions
- Ensuring there is sufficient time available to complete the task
- Modifying the design of the task to make it easy and intuitive
- Implementing simple checklists and clear reminders
- Adding a second cross-check of critical steps
- Introducing warnings and/or alarms
If an error was due to a mistake, improvement actions might include:
- Retraining workers on the task and determine why the initial training was not effective
- Introducing job aids, flow charts, standard work guides, and schematics
- Planning for all relevant non-routine situations
- Developing drills for potential unexpected situations
“ By focusing on identifying and addressing errors due to human factors, some pharma companies have seen a 30-40% reduction in error rates ”
In contrast, if the errors were determined to be intentional actions, the solutions may include:
- Eliminating reasons to cut corners by job redesign, removing unnecessary rules and inconvenient requirements, removing unrealistic targets, ensuring workloads are realistic, and removing adverse environmental factors
- Simplifying the work instructions
- Updating the procedure to reflect actual practice
- Improving risk perception, promote understanding and raise awareness of whys and consequences, clarify the criticality of the task, and explain the impact of not performing the task in the manner required
- Embedding warnings in procedure
- Increasing likelihood of getting caught
- Improving supervision
- Improving attitudes, active workforce involvement, encourage reporting of violations, and make non-compliance socially unacceptable
Key to identifying the cause of the human error or violation – thus ensuring the most appropriate action from the list above is taken – is to ensure that any root cause investigation into an error is thorough and completed in an open, non-blame culture. A blame-based workplace culture can have a negative effect by deterring error reporting and preventing further investigation and remediation. Questions such as those shown in Figure 3 can facilitate identifying the true root cause when a human error or violation is suspected.
Rather than waiting until something goes wrong and then investigating, a more proactive approach is to conduct a study of the human factors related to a specific analytical procedure. By mapping out in detail all the steps involved in carrying out an analysis and observing the analyst performing these steps, it is possible to identify where opportunities for human error exist. A risk assessment can then be performed, and steps taken to put in place mitigating actions for those areas of highest risk.

Error Due to Machine (Instrumentation)
Errors due to equipment issues can become significant if equipment isn’t maintained and replaced at appropriate intervals. To mitigate the risks of an ageing instrumentation base, many organisations look to new technology that can better keep pace with lab throughput, while also serving analytical needs and compliance demands. If capital investment for new instrumentation is limited, then it’s important to protect existing systems with tailored service plans that include timely replacement of worn instrument components with original equipment manufactured (OEM) parts. Partnering with the right instrumentation service vendor is the best way to reduce costs, while also maintaining and supporting lab operations.
Mitigation Strategies
Since instrument errors are systematic errors that have a clear cause and can be readily identified, they can and should be prevented. Actions to mitigate the risk of instrument-related errors include:
- Documenting and recording instrument specifications
- Scheduling regular performance checks
- Completing preventive maintenance tasks
- Performing calibration and qualification, scheduling, and measurements
- Providing training on instrumentation
- Testing to ensure a thorough understanding of operator instructions
- Using data trending to continuously monitor performance and facilitate predictive maintenance
Error Due to Materials
In the lab, materials may include reagents, standards, and instrument-related consumables such as vials, columns, GC liners, etc. To maintain quality assurance and control, lab want to avoid variability in the quality and consistency of materials. That requires minimising any changes to reliable suppliers, testing for quality, and avoiding contamination of supplies.
Mitigation Strategies
Again, prevention is the best method to avoid these types of errors. These strategies may include:
- Performing strict quality testing of materials
- Integrating controls for the administration of the materials
- Limiting variability in acquisition, transportation, and/or storage conditions
- Monitoring environmental conditions, particularly temperature and humidity
- Using standardised management tools
- Documenting all packaging and labels
- Removing, segregating, and/or destroying all expired materials
Conclusion
Risk assessments used to proactively identify potential sources of error, as well as thorough root cause investigations, can help significantly to reduce the prevalence of errors in pharma QC labs. Further, preemptive planning, managing, training, improved workflows, and investment in support services can help manufacturers respond to any errors with an appropriate course of action, thus minimising the potential impact.
Substantial improvements are now being realised across the industry as organisations strive to implement a culture of quality, set targets around error reduction, and update ageing analytical infrastructure. Significant advantages will undoubtedly be realised in the coming years with the development of intelligent technologies and automation that can further mitigate the risk of human errors and unpredicted instrument-related failures.

Dr Stephanie N Harden is the manager of Waters’ global small molecule pharmaceutical market segment, with over 20 years of experience in product and segment marketing. Stephanie has a passion for enabling world-class pharmaceutical development and has been instrumental in helping customers to transform the way their organisations operate. She is a highly cited author with a PhD in Chemistry from the University of Bristol, UK.

Dr Phil Nethercote, Independent Pharmaceutical Industry Consultant. Prior to his retirement, Phil Nethercote was the Analytical Head of GSK’s Manufacturing Division. He played a lead role in reducing lab errors across GSK’s manufacturing sites both through the development and introduction of analytical quality by design principles and through implementing processes to identify and address lab errors due to human factors. He now supports pharmaceutical organisations as an independent consultant.
Innovations in Pharmaceutical Technology (IPT)
IPT provides a platform for cutting-edge ideas, concepts, and developments shaping the future of pharmaceutical R&D.